

GEO生信数据挖掘(八)富集分析(GO 、KEGG、 GSEA 打包带走)-CSDN博客
| 阿里云国内75折 回扣 微信号:monov8 |
| 阿里云国际,腾讯云国际,低至75折。AWS 93折 免费开户实名账号 代冲值 优惠多多 微信号:monov8 飞机:@monov6 |
第六节我们使用结核病基因数据做了一个数据预处理的实操案例。例子中结核类型包括结核潜隐进展对照和潜隐四个类别。第七节延续上个数据进行了差异分析。 本节对差异基因进行富集分析。
目录
数据展示
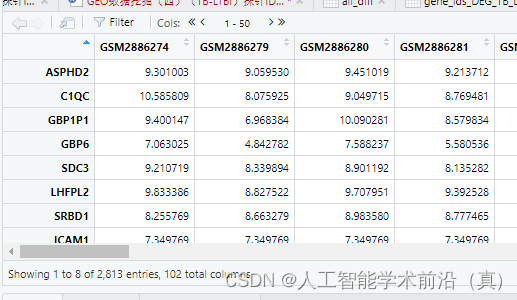
差异基因计算完毕的指标如下图所示

差异基因筛选后表达矩阵
GO富集分析 -对基因名称映射基因ID
加载数据
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#+&&&&&&&&&&&&&&&&&&加载数据&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
load( "DEG_TB_LTBI_step13.Rdata")
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
#+&&&&&&&&&&&&&&&&&&加载数据&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
library(clusterProfiler)
library(org.Hs.eg.db)
#增加基因名
all_diff$SYMBOL=rownames(all_diff)
#基因名称转换注释
gene_ids_DEG_TB_LTBI = bitr(geneID = rownames(dataset_TB_LTBI_DEG),fromType="SYMBOL",toType = c("ENTREZID","ENSEMBL","SYMBOL"),OrgDb = 'org.Hs.eg.db',drop =TRUE)
#合并 增加logFC 为后续GSEA富集分析所需数据准备
gene_ids_DEG_TB_LTBI <- merge(gene_ids_DEG_TB_LTBI,all_diff,by="SYMBOL")
#观察
dim(gene_ids_DEG_TB_LTBI)
head(gene_ids_DEG_TB_LTBI)
#获取基因ID ENSEMBL
gene_ENSEMBL <- gene_ids_DEG_TB_LTBI$ENSEMBL
gene_ENTREZID <- gene_ids_DEG_TB_LTBI$ENTREZID
gene_SYMBOL<- gene_ids_DEG_TB_LTBI$SYMBOL
经过映射2813个差异基因得到2551个基因ID,下图为三种不同形式的基因名称富集分析时按需进行转换。

GO富集分析 -从org.Hs.eg.db库中去匹配基因
#Go富集分析从库中去匹配
go <- enrichGO(gene_SYMBOL,OrgDb = org.Hs.eg.db, ont='ALL',pAdjustMethod = 'BH',pvalueCutoff = 0.05, qvalueCutoff = 0.2,keyType = 'SYMBOL')#进行GO富集确定P值与Q值得卡值并使用BH方法对值进行调整。
#查看富集结果
dim(go)
#导出GO富集的结果
write.csv(go,file="go1.csv")绘制气泡图
#绘制气泡图
pdf(file="15aGO富集分析step15.pdf", width = 9, height = 6)
dotplot(go,showCategory=20,label_format = 80)#气泡图
dev.off()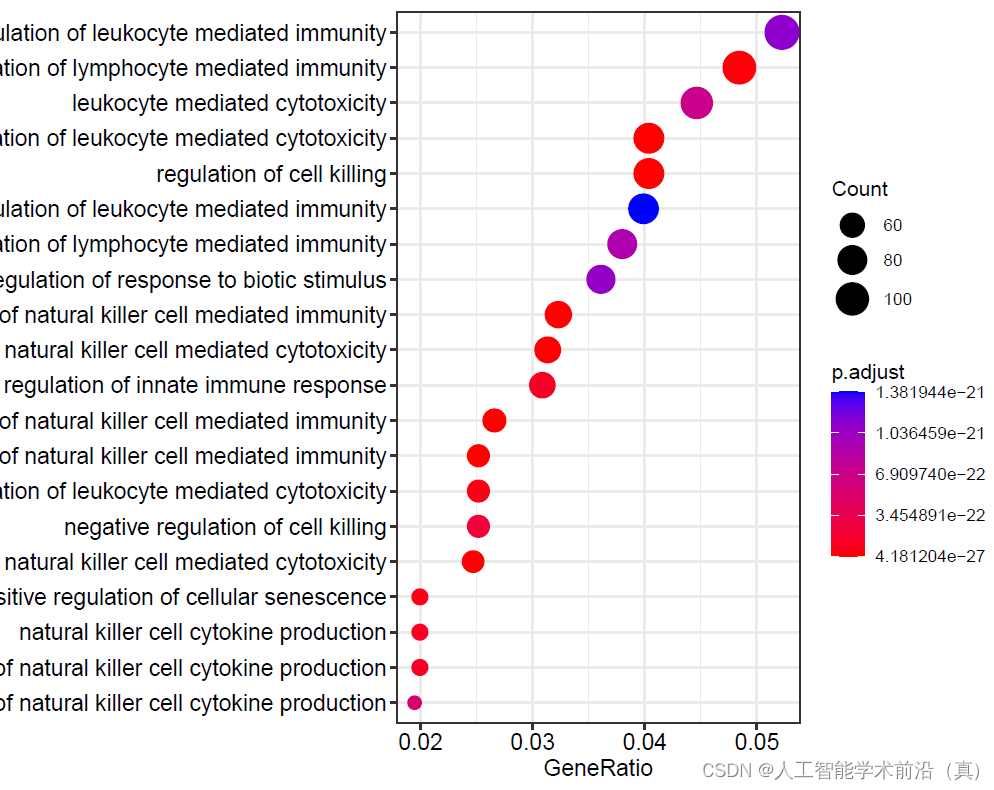
三种不同类别的合并的气泡图#CC细胞组件MF分子功能BP生物学过程
pdf(file="15bGO富集分析三组step15.pdf", width = 9, height = 6)
#CC细胞组件MF分子功能BP生物学过程
goCC <- enrichGO(gene = gene_ENTREZID, #基因列表(转换的ID)
keyType = "ENTREZID", #指定的基因ID类型默认为ENTREZID
OrgDb=org.Hs.eg.db, #物种对应的org包
ont = "CC", #CC细胞组件MF分子功能BP生物学过程
pvalueCutoff = 0.05, #p值阈值
pAdjustMethod = "fdr", #多重假设检验校正方式
minGSSize = 1, #注释的最小基因集默认为10
maxGSSize = 500, #注释的最大基因集默认为500
qvalueCutoff = 0.2, #q值阈值
readable = TRUE) #基因ID转换为基因名
goBP <- enrichGO(gene_ENTREZID,OrgDb = org.Hs.eg.db, ont='BP',pAdjustMethod = 'BH',pvalueCutoff = 0.05, qvalueCutoff = 0.2,keyType = 'ENTREZID')
goMF <- enrichGO(gene_ENTREZID,OrgDb = org.Hs.eg.db, ont='MF',pAdjustMethod = 'BH',pvalueCutoff = 0.05, qvalueCutoff = 0.2,keyType = 'ENTREZID')
#通过ggplot2将BP、MF、CC途径的富集结果挑选前8条绘制在一张图上
barplot(go,label_format=100, split="ONTOLOGY")+ facet_grid(ONTOLOGY~.,scale="free")
dev.off()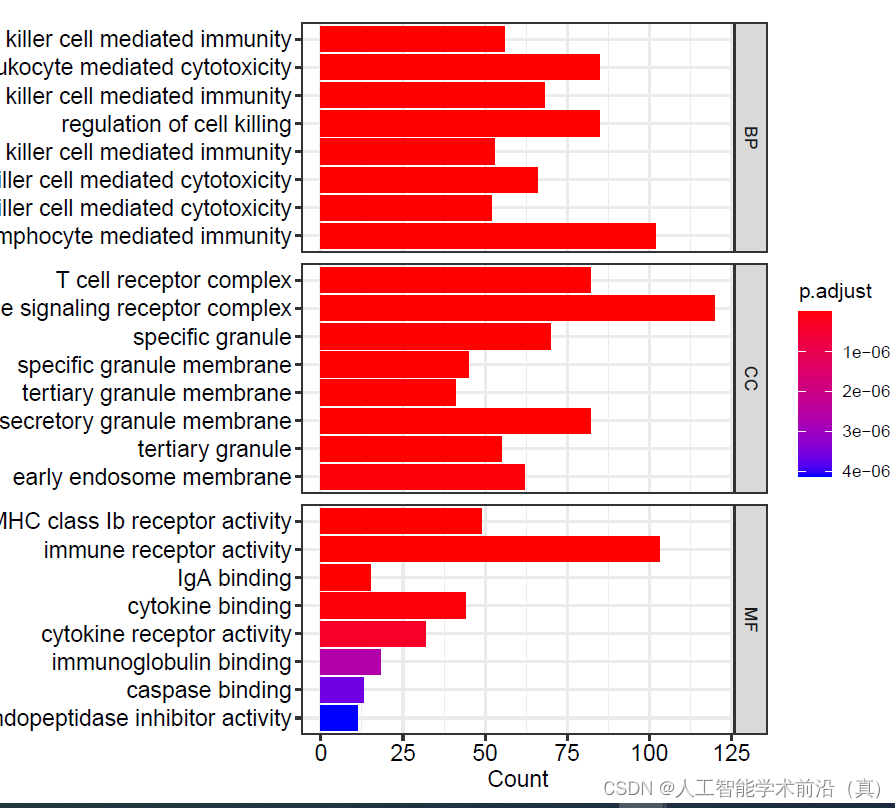
KEGG富集分析 不详细讲了看注释
#+=================================================================
#============================================================
#+========KEGG富集分析 气泡图step16===================
#+==========================================
#+================================
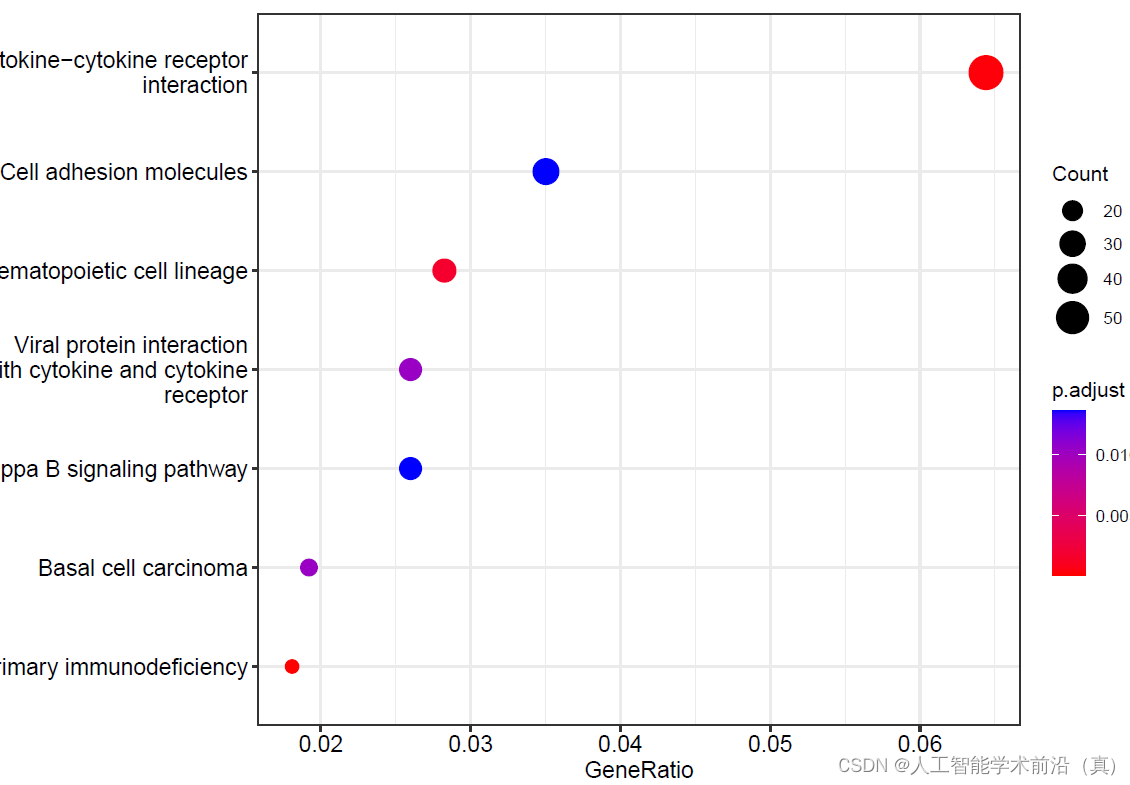
#KEGG富集分析
pdf(file="16KEGG富集分析step16.pdf", width = 9, height = 6)
kegg<- enrichKEGG(gene = gene_ENTREZID, #基因列表(ENTREZID ID: 54490,51144,31,3906)
organism = "hsa", #物种
keyType = "kegg", #指定的基因ID类型默认为kegg
minGSSize = 3,
maxGSSize = 500,
pvalueCutoff = 0.05,
pAdjustMethod = "fdr", # pAdjustMethod = 'BH'
qvalueCutoff = 0.02)
#观察
dim(kegg)
#绘制气泡图
dotplot(kegg)
dev.off()
#kegg 增加可读性对基因ID 转基因名
kegg_enrich_results <- DOSE::setReadable(kegg,
OrgDb="org.Hs.eg.db",
keyType='ENTREZID') #ENTREZID to gene Symbol
#保存kegg结果
write.csv(kegg_enrich_results@result,'KEGG_gene_up_enrichresults.csv')
#save(kegg_enrich_results, file ='KEGG_gene_up_enrichresults.Rdata')
##查看与选择所需通路
kegg_enrich_results@result$Description[1:10] #查看前10通路
###选择所需通路的ID号
i=1
select_pathway <- kegg_enrich_results@result$ID[i] #选择所需通路的ID号
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
save(gene_ids_DEG_TB_LTBI,go,keggfile ="15_gene_ids_DEG_TB_LTBI.Rdata")
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
GSEA 富集分析
#+=================================================================
#============================================================
#+========GSEA 富集分析 气泡图step17===================
#+==========================================
#+================================
# GSEA 分析
#需要把多个方法取并集
#该方法的输入需要基因和 logFC 排序后的结果
#不同方法 相同基因的的logFC值不一样直接保留第一个重复基因
library(stringr)
## 去重 #去除NA值
dim(gene_ids_DEG_TB_LTBI)
colnames(gene_ids_DEG_TB_LTBI)
gene_list_df = gene_ids_DEG_TB_LTBI[,c('ENTREZID','logFC')]
gene_list_df_na <- na.omit(gene_list_df)
gene_ids_TB_LTBI_distinct <- dplyr::distinct(gene_list_df_na,ENTREZID,.keep_all=TRUE)
dim(gene_ids_TB_LTBI_distinct)
gene_list=gene_ids_TB_LTBI_distinct$logFC #提取logFC列
names(gene_list)=gene_ids_TB_LTBI_distinct$ENTREZID #加上ENTREZID
gene_list_gsea = sort(gene_list, decreasing = T) #降序排列
gsea_KEGG <- gseKEGG(gene_list_gsea,
organism = "hsa",
keyType = "kegg")
gsea_KEGG_d <- as.data.frame(gsea_KEGG)
gsea_KEGG_d
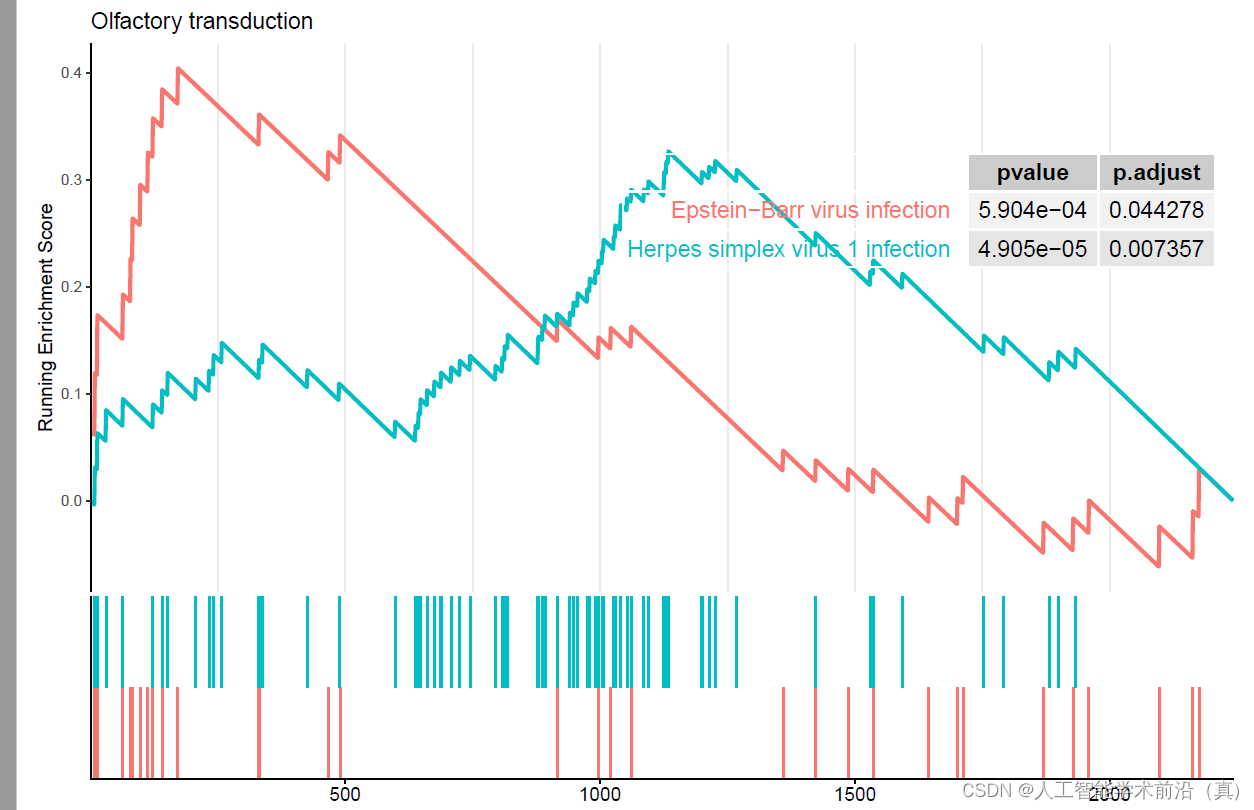
#path 为需要展示的pathway id这里展示的是enrichment score最高的4条通路
t_index=order(gsea_KEGG_d$enrichmentScore,decreasing = T)
path=rownames(gsea_KEGG[t_index,]) #选择展示的 pathwayrownames(gsea_KEGG[t_index,]) [1:4]
#作图
pdf(file="17GSEA富集分析step17.pdf", width = 9, height = 6)
gseaplot2(gsea_KEGG,
path,
subplots = 1:2, #展示前2个图
pvalue_table = T, #显示p值
title = "Olfactory transduction", #设置title
base_size = 10, #字体大小
color="red") #线条颜色可选
dev.off()
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
save(gene_ids_DEG_TB_LTBI,go,kegg,gene_list_gsea,gsea_KEGG,file ="17_gene_ids_DEG_TB_LTBI.Rdata")
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
更多复杂的图关联网络图、八卦图 、弦图
参考 最全的GO, KEGG, GSEA分析教程(R),你要的高端可视化都在这啦[包含富集圈图] - 糖糖家的老张的文章 - 知乎 https://zhuanlan.zhihu.com/p/377356510
原文链接https://blog.csdn.net/qq_50898257/article/details/120588222
#+=================================================================
#============================================================
#+========富集分析 更多的图step18===================
#+==========================================
#+================================
library(clusterProfiler)
library(enrichplot)
#+富集基因与所在功能集/通路集的关联网络图
enrichplot::cnetplot(go,circular=FALSE,colorEdge = TRUE)#基因-通路关联网络图
enrichplot::cnetplot(kegg,circular=FALSE,colorEdge = TRUE)#circluar为指定是否环化基因过多时建议设置为FALSE
GO2 <- pairwise_termsim(go)
KEGG2 <- pairwise_termsim(kegg)
enrichplot::emapplot(GO2,showCategory = 50, color = "p.adjust", layout = "kk")#通路间关联网络图
enrichplot::emapplot(KEGG2,showCategory =50, color = "p.adjust", layout = "kk")
write.table(kegg$ID, file = "KEGG_IDs.txt", #将所有KEGG富集到的通路写入本地文件查看
append = FALSE, quote = TRUE, sep = " ",
eol = "\n", na = "NA", dec = ".", row.names = TRUE,
col.names = TRUE, qmethod = c("escape", "double"),
fileEncoding = "")
#打印几条通路名称看看
kegg$ID[1:3]
#打开浏览器观察通路
browseKEGG(kegg,"hsa04660")#选择其中的hsa05166通路进行展示
#富集弦图
genedata<-data.frame(ID=gene_ids_DEG_TB_LTBI$SYMBOL ,logFC=gene_ids_DEG_TB_LTBI$logFC)
write.table(go$ONTOLOGY, file = "GO_ONTOLOGYs.txt", #将所有GO富集到的基因集所对应的类型写入本地文件从而得到BP/CC/MF各自的起始位置如我的数据里是121032410
append = FALSE, quote = TRUE, sep = " ",
eol = "\n", na = "NA", dec = ".", row.names = TRUE,
col.names = TRUE, qmethod = c("escape", "double"),
fileEncoding = "")
'''
根据计算出的go 文件数量调整
'''
GOplotIn_BP<-go[1:178,c(2,3,7,9)] #提取GO富集BP的前10行,提取ID,Description,p.adjust,GeneID四列
GOplotIn_CC<-go[179:194,c(2,3,7,9)]#提取GO富集CC的前10行,提取ID,Description,p.adjust,GeneID四列
GOplotIn_MF<-go[195:209,c(2,3,7,9)]#提取GO富集MF的前10行,提取ID,Description,p.adjust,GeneID四列
library(stringr)
GOplotIn_BP$geneID <-str_replace_all(GOplotIn_BP$geneID,'/',',') #把GeneID列中的’/’替换成‘,’
GOplotIn_CC$geneID <-str_replace_all(GOplotIn_CC$geneID,'/',',')
GOplotIn_MF$geneID <-str_replace_all(GOplotIn_MF$geneID,'/',',')
names(GOplotIn_BP)<-c('ID','Term','adj_pval','Genes')#修改列名,后面弦图绘制的时候需要这样的格式
names(GOplotIn_CC)<-c('ID','Term','adj_pval','Genes')
names(GOplotIn_MF)<-c('ID','Term','adj_pval','Genes')
GOplotIn_BP$Category = "BP"#分类信息
GOplotIn_CC$Category = "CC"
GOplotIn_MF$Category = "MF"
BiocManager::install('GOplot')
library(GOplot)
circ_BP<-GOplot::circle_dat(GOplotIn_BP,genedata) #GOplot导入数据格式整理
circ_CC<-GOplot::circle_dat(GOplotIn_CC,genedata)
circ_MF<-GOplot::circle_dat(GOplotIn_MF,genedata)
chord_BP<-chord_dat(data = circ_BP,genes = genedata) #生成含有选定基因的数据框
chord_CC<-chord_dat(data = circ_CC,genes = genedata)
chord_MF<-chord_dat(data = circ_MF,genes = genedata)
'''
> chord_CC<-chord_dat(data = circ_CC,genes = genedata)
Error in `[<-`(`*tmp*`, g, p, value = ifelse(M[g] %in% sub2$genes, 1, :
subscript out of bounds
我去检查了go和genelist的数据结构发现genelist里的gene用的是gene名而go里的基因用的是基因ID不一样了所以跑不出结果所以我把genelist的gene换成了基因ID就能跑出来了。
作者ff的小世界勿扰
链接https://www.jianshu.com/p/ee4012fd253f
来源简书
著作权归作者所有。商业转载请联系作者获得授权非商业转载请注明出处。
'''
#可以画 数量太多了
GOChord(data = chord_BP,#弦图
title = 'GO-Biological Process',space = 0.01,#GO Term间距
limit = c(1,1),gene.order = 'logFC',gene.space = 0.25,gene.size = 5,
lfc.col = c('red','white','blue'), #上下调基因颜色
process.label = 10) #GO Term字体大小
GOChord(data = chord_CC,title = 'GO-Cellular Component',space = 0.01,
limit = c(1,1),gene.order = 'logFC',gene.space = 0.25,gene.size = 5,
lfc.col = c('red','white','blue'),
process.label = 10)
GOChord(data = chord_MF,title = 'GO-Mollecular Function',space = 0.01,
limit = c(1,1),gene.order = 'logFC',gene.space = 0.25,gene.size = 5,
lfc.col = c('red','white','blue'),
process.label = 10)
'''
Warning messages:
1: Using size for a discrete variable is not advised.
2: Removed 15 rows containing missing values (`geom_point()`).
'''富集分析完毕
回顾我们用到方法差异分析后进行富集分析理论基础实际上就是简单的找不同分析。
实际应用种由于基因之间存在关联另一套分析理论考虑的是基因之间的相互作用下节我们来看非常火的WGCNA 共表达加权网络进行基因分析。