

论文解读:DNA 5-甲基胞嘧啶检测和甲基化分相 PacBio 循环共识测序
| 阿里云国内75折 回扣 微信号:monov8 |
| 阿里云国际,腾讯云国际,低至75折。AWS 93折 免费开户实名账号 代冲值 优惠多多 微信号:monov8 飞机:@monov6 |
Title:DNA 5-methylcytosine detection and methylation phasing using PacBio circular consensus sequencing
期刊:nature communications
中科院分区:Q1
影像因子:16.6↓ 1.094
Github:
文章链接: https://doi.org/10.1038/s41467-023-39784-9
摘要
长单分子测序技术,如 PacBio 环形共识测序(CCS)和纳米孔测序,在生物医学领域具有优势检测 CpGs (5mcpg)中的 DNA 5-甲基胞嘧啶,特别是在重复的基因组区域。然而,使用 PacBio CCS 检测 5mcpg 的现有方法准确性和鲁棒性较差。在这里,我们提出了 ccsmeth,一种使用 CCS 读取检测 DNA 5mcpg 的深度学习方法。 我们使用 PacBio CCS 对一个人类样本的聚合酶链反应处理和 m.s sssi 甲基转 移酶处理的 DNA 进行测序,以进行训练。使用长(≥10 Kb) CCS 读取,ccsmeth 在单分子分辨率下 5mCp G 检测的准确度为 0.90,曲线下面积为 0.97。在全基因组水平,ccsmeth 与亚硫酸氢盐测序和纳米孔测序仅使用 10× reads就实现了>0.90 的相关性。此外,我们开发了 Nextflow 管道 ccsmethphase,使 用 CCS 读取检测单倍型感知甲基化,然后对中国家庭三组进行测序以验证它。ccsmeth 和 ccsmethphase 是检测 DNA 5-甲基胞嘧啶的可靠和准确的工具。
引言
5-甲基胞嘧啶(5mC)是 DNA 甲基化最常见的形式,参与调节许多生物过程 1。在人类中, 大多数 5mc 发生在 CpG 位点, 与胚胎发育、疾病和衰老 2,3 相关。亚硫酸氢盐测序(BS-seq)是目前最广泛使用的分析 5mC 甲基化的方法。4 在亚硫酸处理的基因组 5DNA 中, 未甲基化的胞嘧啶转化为尿嘧啶,而甲基化的胞嘧啶不变。因此,一段 DNA的甲基化状态可以在单核苷酸分辨率下得到。然而, 亚硫酸氢盐处理会破坏 DNA, 进一步导致 DNA 降解和测序丧失多样性 6。近年来, 人们开发了两种不含亚硫酸盐的方法,即 10 -11 易位辅助吡啶硼烷测序(7 TAPS)和酶促甲基测序(8 EM-seq),这两种方法都比 BS-se q 具有更均匀的覆盖范围和更高的独特定位率。与 BS- seq 一样, TAPS 和 EM-seq 既可以用于短读段测序,也可以用于长读段测序。9– 11 然而, 所有这些方法都需要额外的实验室技术, 这进一步导致了额外的测序成本。
两个主要的长读测序技术, PacBio 单分子实时( SMRT)测序和纳米孔测序牛津纳米孔技术(ONT),可以直接测序原生 DNA,无需 PCR 扩增。 12,13DNA 碱基修饰改变了 SMRT 测序中的聚合酶动力学,并影响了纳 米孔测序中修饰碱基附近的电流信号 13。因此, DNA 碱基修饰可以 直接从 SMRT 和纳米孔测序的天然 12,13DNA 读取中检测到,而无需额 外的实验室技术。对于纳米孔测序, 5mC 检测的计算方法要么应用 统 计测试来 比较原生 DNA 读 取 的当 前信号与 未修改 的对 照 (Tombo14),要么使用预训练的隐马尔可夫模型(nanopolish15)和深度 神经网络模型(Megalodon16,Deep Signal17),而不使用对照数据集。先前的研究表明 18,19 , 使用预训练模型的方法对人类纳米孔读取的 DNA 5mC 检测具有很高的准确性。
SMRT 测序中的脉冲信号与聚合反应发生 13,20 的核苷酸相关,包括脉冲间隔时间(IPD)和脉冲宽度(PW)。IPD 表示两个连续测序碱基之间的时间间隔。PW 表示碱基被测序 20 的持续时间。除了被测序的核苷酸,碱基修饰也会影响脉冲信号。利用修改碱基和未修改碱基之间脉冲信号的差异, 开发了从 SMRT 数据中检测 5mC 和其他碱基修改的方法。21 然而 12,13,由于信噪比较低,使用早期版本的 SMRT 数据可靠地调用 5mC 需要高的读取覆盖率(高达 250x)。基于脊椎动物中未甲基化的 CpGs 通常分布在较长的低甲基化区域, Suzuki 等人提出了 AgIn, 通过结合 SMRT 数据中邻近 CpGs 的 IPD特征,提高了 5mCpG 检测的可信度。22 最近, 提出了 PacBio 循环共识测序(CCS)技术,该技术利用单个零模波导(ZMW)中的循环模板 23 生成的子读段来调用高精度的共识序列(CCS/HiFi 读段)。利用新的 CCS 技术, Tse 等人开发了一种基于卷积神经网络(CNN)的方法, 称为整体动力学模型(HK 模型), 用于人类全基因组 5mCpG 检测。24 对于 CCS 序列, HK 模型首先计算每个碱基的平均 IPD ,还使用 15 Kb 插入长度的 PacBio CCS 和 BS-seq 对人类男性样本 SD0651_P1 进行了测序。在 hg23,26 002 和 chm1327 的测序数据集和公开数据集上进行的实验表明, ccsmeth 在读取水平检测 5mCpG 的精度高于 HK 模型和 primrose。ccsmeth 还在全基因组位点水平上实现了与 BS- seq 和纳米孔测序的高度相关性。结果表明, ccsmeth 可以在读取水平和位点水平上准确检测长(≥ 10 Kb) CCS 读取的 Cp Gs 的甲基化状态。
等位基因特异性甲基化(ASM)既发生在印记区域,也发生在非印记区域, 与复杂疾病和癌症 28,29 相关。30 最近的研究表明, PacBio CCS 测序和纳米孔测序都可以用于单倍型感知的基因组组装、变异召唤 31 和甲基 32,3334– 37 化相位。在这里,通过改进 ccsmeth 的 5mCpG检 测 , 我 们 进 一 步 开 发 了 一 个 名 为 ccsmethphase38 的 Nextflowpipeline,使用 CCS 读取检测单倍型感知甲基化。我们还使用 PacBioCCS 对一个中国家系进行测序以验证 ccsmephase。测试数据集的结果表明, ccsmephase 可以准确地检测全基因组等位基因特异性甲基化。此外, 我们证明了 PacBio CCS 现在是一种全面而准确的 5mCpG 检测和甲基化分期技术,即使在重复的基因组区域也是如此。
结果
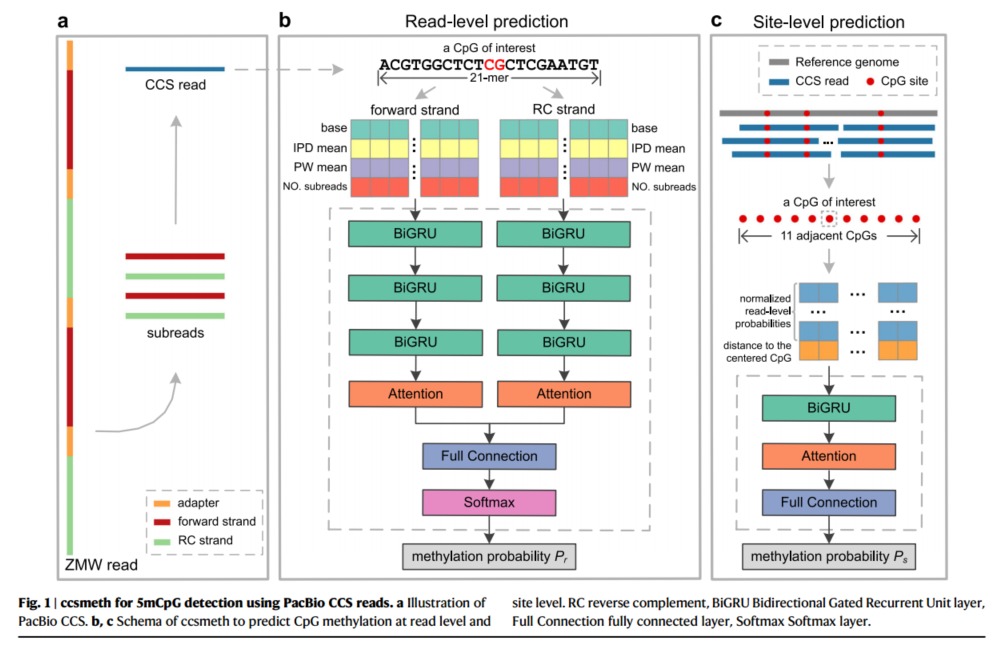
ccsmeth 算法用于 5mCpG 检测递归神经网络(Recurrent neural network, RNN)和注意机制是在自然语言处理中应用广泛的人工神经网络。39,40RNN 和注意机制都被应用于PW 值,然后将 CCS 序列的亚 reads 与参考基因组进行比对。然后, 碱基修饰检测中。16,17,41 在这里, 我们提出了 ccsmeth, 这是一种由双对于 CCS 读取中的每个 CpG 位点, HK 模型将平均 IPD 值、平均PW 值和 CpG 周围的序列上下文组织成一个特征矩阵。最后, HK向 gru 和 Bahdanau 注意 42 网络组成的深度学习方法,用于检测 Pac- Bio CCS读取的 Cp G43 甲基化。ccsmeth 旨在预测 CpGs 在读取水平和模型将特征矩阵输入到基于 cnn 的模型中, 得到 CpG 的甲基化概率。 位点水平上的甲基化状态。对于一个目标 CpG, ccsmeth 首先预测该24HK 模型在读取水平(即单分子分辨率)检测 5mCpG 的灵敏度和特异性均达到 90%以上。然而, HK 模型需要较高的 CCS 子读深度(一个 CCS 至少需要 20 倍的通过子读深度)才能准确检测 5mCpG,这限制了文库制备中的插入大小, 进一步限制了 CCS 读取的长度。继 HK模型之后,PacBio 提出了另一种基于 cnn 的方法 primrose,据称该方法对长 PacBio CCS读取的 5mCp G检测 25 具有 85%的读取级准确率 。 此 外 , PacBio在 pb- Cp G-tools (https://github.com/Pacific Bio sciences/ pb- Cp G-tools) 中 提 供 了 primrose 的伴侣脚本来预测 CpGs 的位点水平甲基化频率。与 AgIn类似, 对于基因组中的每个 CpG, pb- Cp G-tools 将报春花预测的 Cp G 及其邻近 Cp G 的读级甲基化概率 22 组织为特征。然后, p b- Cp G工具将这些特征输入到基于 cnn 的模型中, 以获得目标 CpG的预测甲基化频率。在这项研究中, 我们提出了 ccsmeth,这是一种深度学习方法,通过使用 PacBio CCS 读取的动力学特征(ipd 和 PWs)来检测 DNA 5mcpg。ccsmeth 利用双向门控循环单元(GRU)和注意力神经网络, 在读取水平和全基因组位点水平检测 CpGs 的甲基化状态。为了评估 ccsmeth 的性能,我们使用插入长度为 10 Kb 的 PacBio CCS对扩增和m.s ssi 处理的人样本 NA12898 的 DNA 进行测序。我们CpG 在一个 read(即在单分子分辨率, read 水平)中的甲基化概率(或二元甲基化状态), 然后总结这些 read-level 甲基化状态,得到其在目标基因组中的甲基化频率(位点水平)(图 1,Methods)。在 CCS读取的生成过程中, CCS读取的正向和反向补链中每个碱基的 IPD 和 PW 值从相应的子读取中取平均值(图 1a)。为了预测 CpG 的读级甲基化状态,ccsmeth 提取了一个 21-mer 序列上下文,其中包括位于中心的 CpG本身,以及每个碱基的动力学信息(平均 IPD、平均 PW 和覆盖的亚读的数量)。由于人类 CpG甲基化大多是对称的, ccsmeth 从正向和反向补体链构建了对称 CpG对的两个特征矩阵 44(图 1b)。在处理特征矩阵之后, ccsmeth 输出一个读取级甲基化概率 Pr (Pr 2½0,1 ?)。
在调用位点水平的甲基化之前, CCS 读数应该与参考基因组对齐。在 ccsmeth 中, 我们提供了两种模式来推断 CpG的位点级甲基化频率:计数模式和模型模式(方法,补充图 1)。在计数模式中, 基于读取级 甲基化概率,每个读取中 CpG 的二进制甲基化状态(0 为未甲基化, 1为甲基化)由概率截止值(默认为 0.5)设置。然后, 通过计算预测 CpG 被甲基化的读取的数量和映射到 CpG 的读取的总数来计算甲基化频 率。在 ccsmeth 的模型模式中,我们利用邻近 CpG的读级甲基化概率,以类似于 pb- cpg 工具的方式增加位点级甲基化检测的置信度。具体 来说,对于一个目标 CpG,该 CpG 及其相邻的 10 个 CpG 的读级甲基化概率,以及所有 11 个 CpG 到目标 CpG 的距离(碱基对)被组织成 一个特征矩阵。首先将特征矩阵输入到 BIGRU 层中来捕捉相邻 CpG 之间的正向和反向相互作用。然后应用一个关注层来优化每个相邻 CpG 的权重, 这使得模型能够专注于最相关的交互。 10-25 Kb23。因此, 我们进一步使用三个人类样本(NA12898、HG002甲基化概率 Ps (P2s ½0,1 ?)最终作为目标 CpG 的甲基化频率输出(图 1c,补充图 1)。
ccsmeth 在单分子分辨率下精确检测 CpG 甲基化为了在读取水平(即单分子分辨率)上评估 ccsmeth,我们首先使用三组 CCS 数据集(M01&W01, M02&W02, M03&W03) 在不同版本的和 SD0651_P1)的长(≥ 10 Kb) CCS reads 进行读取级评估(方法,补充表 2):NA12898 的 CCS reads 使用 NA12898 的 pcr 处理的和 m.ssi 处理的 10 Kb 插入大小的 NA12898 的 DNA 进行测序;HG002 原生 DNA采用三种不同的插入长度(15Kb、20Kb、24Kb)测序, CCS reads 来自 Baid 等 26 人和 Human Pangenome Reference Consortium;23 SD0651_P1的 CCS序列使用 15Kb DNA 插入长度进行测序。这些数据集中 CCS读取的平均亚读深度范围为 7.6× ~ 14.1×(补充表 1)。我们使用与参PacBio 测序仪上使用 m.s sssi 处理和 pcr 处理的人类 DNA 进行测序。 考基因组常染色体对齐的 NA12898 CCS 读取和 HG002 CCS 读取的24 在 m.s sssi 处理的 DNA 中, Cp G甲基转移酶 m.s rsi 使所有 Cp G甲基化,而通过全基因组扩增(WGA)24 制备的 pcr 处理的 DNA 几乎没有甲基化碱基。如补充表 1 所示, M01-03 为 m.s sssi 处理的 DNA样本, W01-03 为 pcr 处理的 DNA 样本。对于每组数据集,我们随机选取 50%甲基化的 reads 和非甲基化的 reads 进行模型训练。剩下的 50%甲基化和非甲基化的读长用于测试。我们使用相同的读数来训练和测试 HK 模型。24Primrose 不提供训练的接口, 所以我们排除了它来进行这三个数据集的比较。如图 2a 所示, ccsmeth 在所有三个数据集上都优于 HK 模型。ccsmeth 在 M01&W01、M02&W02 和 M03&W03 上的精度分别为 0.9232、0.8788 和 0.8765。ccsmeth 模型在三个数据集上的精度分别比 HK 模型高 5.4 %、4.4%和 3.7 %。 ccsmeth 模型在三个数据集上的 auc 均在 0.95 左右或以上,分别比 HK 模型高 3.3%、3.4%和 2.6%。来自 Tse 等 24 人的三个 CCS 数据集的读取长度都小于 10Kb,而实际使用的 CCS读取通常在一个 SMRT 细胞来训练 ccsmeth 的读取水平模型(Methods)。使用与 chrX 对齐的 NA12898 CCS 读取, HG002 CCS 读取的 6 个 SMRT 细胞和 SD0651_P1 CCS 读取进行测试(方法)。我们在相同的测试数据上运行带内置模型的樱草花(primrose) 进行比较。 如图 2b 所示, ccsmeth 在 5 个数据集上的准确率 为 0.8721-0.9062 , 灵敏度 为 0.8621-0.8903, 特异性为 0.8765-0.9220,auc 为 0.9464-0.9682,均高于 primrose。特别是在 15 Kb、20 Kb 和 24 Kb 插入长度的 HG002 CCS 序列上, ccsmeth 的准确度和特异性比 primrose 高 4%以上, 特异性比 primrose 高 7%以上。为了将 ccsmeth 与 HK 模型进行比较,我们从 NA12898 数据集和三种 HG002 插入大小(15 Kb, 20 Kb, 24 Kb)的数据集中对 100 K ZMW 读取进行了抽样,因为 HK 模型在大数据集上非常耗时。结果表明, HK 模型在 HG002 数据集上的精度与报春花相当。HK 模型和报春花的准确性都低于 ccsmeth(补充图 2)。除了广泛评估 ccsmeth 基因组外,我们还在特定的基因组背景和区域中评估 ccsmeth, 以探索 ccsmeth 的表现是否与任何基因组特征相关(补充注 1)
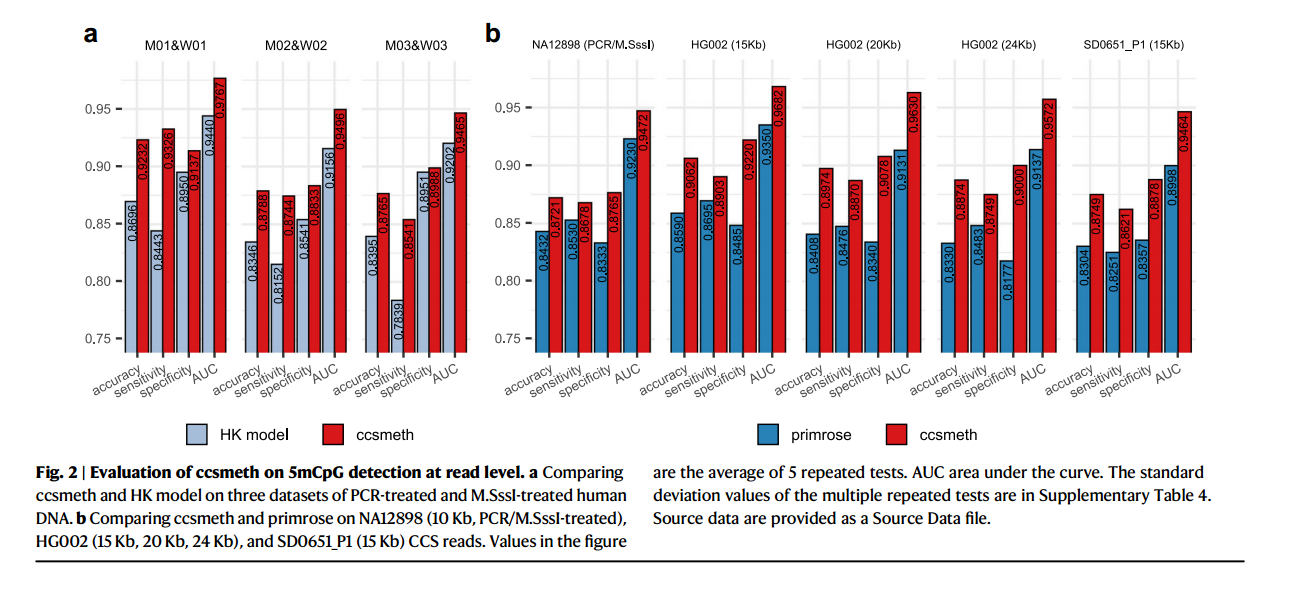
图 2 |读取水平 ccsm e th 对 5mC p G 检 测的评价。 在 pcr 处理和 m.s sssi 处理的三组人类DNA 数据集上比较 ccs meth 和HK 模型。b 在NA12898 (10 Kb, PCR/ m.s sssi 处理)、 HG002 (15 Kb, 20 Kb, 24 Kb)和SD0651_P 1 (15 Kb) CCS 上比较ccs meth 和报春花。图中的值 为 5 次重复测试的平均值。AUC 曲线下面积。多次重复试验的标准差值见补充表 4。源数据作为源数据文件提供。
在所有测试地区都优于报春花。ccsmeth 在 CpG 密度高的区域具有较高的精度,而在成因间区、CpG 海岸、CpG 陆架和一些重复区域具有相对较低的精度。
方法
通过过滤掉模棱两可的调用, ccsmeth 的读级精度可以进一步提高。如补充图 4a 所示, 通过过滤掉甲基化概率接近 0.5 的调用, 可以减少 ccsmeth 的错误调用。我们定义 Δp = |Pr- P 'r |来过滤掉模棱两可的调用,其中 pi 是甲基化概率, P 'r r 是定义为 1- Pr 的未甲基化概率。与modbam2bed 一样, 我们将 Δp 设置为 0.33 进行测试。45 补充图 4b 显示, 当 Δp 设置为 0.33 时, ccsmeth 的准确率提高了 3.5-4.2%, 其中有 8.9-12.8%的呼叫被丢弃。
ccsmeth 在全基因组位点水平准确检测 CpG 甲基化
我们使用 HG002 、 SD0651_P1 和 CHM13 的 CCS 读数来评估 ccsmeth 在站点水平的 5mCpG 检测(方法,补充表 1 和 2)。使用 HG002 CCS读数的 2 个 SMRT 细胞来训练 ccsmeth 的站点水平模型;测试使用 HG002 CCS 读取单元 6 个(15Kb、20Kb、24Kb 三种插入大小各 2 个), SD0651_P1 15Kb 读取 SMRT 单元 2 个, CHM13 20Kb 读取 SMRT 单元 2 个。用于检测的 HG002 (15Kb、20Kb、24Kb)、 SD0651_P1 和 CHM13 CCS reads 的平均基因组覆盖率分别为 25.6×、17.0×、28.4×、19.6×和 16.5×。我们下载了三个人体样本的 BS- seq 和纳米孔 R9.4.1 测序数据作为基准(补充表 3)。在评估 ccsmeth 时, 我们将五个数据集在一定覆盖率下的亚样本读取,并将 ccsmeth 和报春花的位点水平结果与 BS- seq 和纳米孔测序的结果进行比较(图 3a-d)。我们对每个覆盖重复 5 次子样本采样, 并获得用于比较的指标的平均值。结果表明, ccsmeth 和报春花/pb-Cp G- tools 的模型模式与 BS-se q 和纳米孔测序的 Pearson 相关性高于 ccsmeth 和报春花/pb-CpG-tools 的计数模式。同时,通过设置 Δp 为0.33 来过滤歧义调用, ccsmeth 计数模式与 BS-se q 和纳米孔测序的 Pearson 相关性提高了~1-2%( 图 3a-d)。 在所有测试的数据集上, ccsmeth 在两种模式下都比 primrose/pb-Cp G- tools 获得了更高的相关性,特别是在低覆盖率下。例如,使用 10× HG002 15 Kb、20 Kb 和 24 Kb CCS读取, ccsmeth 在模型模式下 primrose/pb-CpG-tools 与 BS- seq 的相关性分别为 0.9198、 0.9083 和 0.9087, 而模型模式下 primrose/pb-CpG-tools 的相关性分别为 0.8864、0.8653 和 0.8696。(图 3)。模型模式下的 ccsmeth 在使用 10× HG002 15 Kb、20 Kb 和 24 Kb CCS reads 时, 与纳米孔测序的相关 性也 分别 为 0.9062 、 0.8967 和 0.8952 , 分别 比模 型模 式下 primrose/pb-CpG-tools 获得的相关性高 4.3%、5.5%和 4.9%(图 3b)。在所有测试的数据集上, ccsmeth 在大多数情况下也比 primrose/pb-CpG- 获得更低的均方根误差(rmse)(补充表 5-12)。
ccsmeth 的模型模式也可以应用于报春花的读级结果。如补充表 5-12 所示, 无论是 BS-se q 还是纳米孔测序, 模型模式下 ccsmeth 的 报春花都比计数模式下 pb- cpg 工具的报春花具有更高的相关性和更低的 rmse。特别是在 HG002 和 CHM13 数据集的低覆盖率(<15 倍)下,模型模式下 ccsmeth 的报春花优于模型模式下 pb-CpG-tools 的报春花(补充表 5-10 和 12)。这些结果进一步证明了 ccsmeth 位点水平模型的有效性。
我们使用 HG002 15 Kb、20 Kb、24 Kb、SD0651_P1 和 CHM13的总 CCS 读数进一步测试 ccsmeth。ccsmeth 使用 HG002 15 Kb、20 Kb 和 24 Kb 数据集的 reads,与 BS-seq 的相关性分别为 0.9463、 0.9271 和 0.9410,与纳米孔测序的相关性分别为 0.9287、0.9127 和0.9240(图 3e)。结合 HG002 的 71.0× CCS总读数, ccsmeth 与 BS-se q和纳米孔测序的相关性分别为 0.9571 和 0.9394(补充图 5, 补充数据1)。ccsmeth 与 SD0651_P1 总读数的相关性为 0.8750,与 CHM13 总读数的相关性为 0.9328(图 3f, g,补充图 6)。ccsmeth 在三个 HG002数据集上的结果也高度相关(相关系数>0.9344), 说明 ccsmeth 具有可重复性(图 3e)。我们进一步使用 71.0× HG002 CCS 读数来探索模型模式在甲基化频率方面更准确地预测哪些 CpG 上下文(补充注释 2 和补充图 7)。我们将 CpG 分为 Gm 和 Gc 两组。Gm 含有的 CpGs 的甲基化频率可以通过模型模式更准确地预测,而 Gc 含有的 CpGs 的甲基化频率可以通过计数模式更准确地预测。我们发现 Gm 中的 CpGs 往往具有非常低(<0.2)或高(>0.8)的甲基化频率。
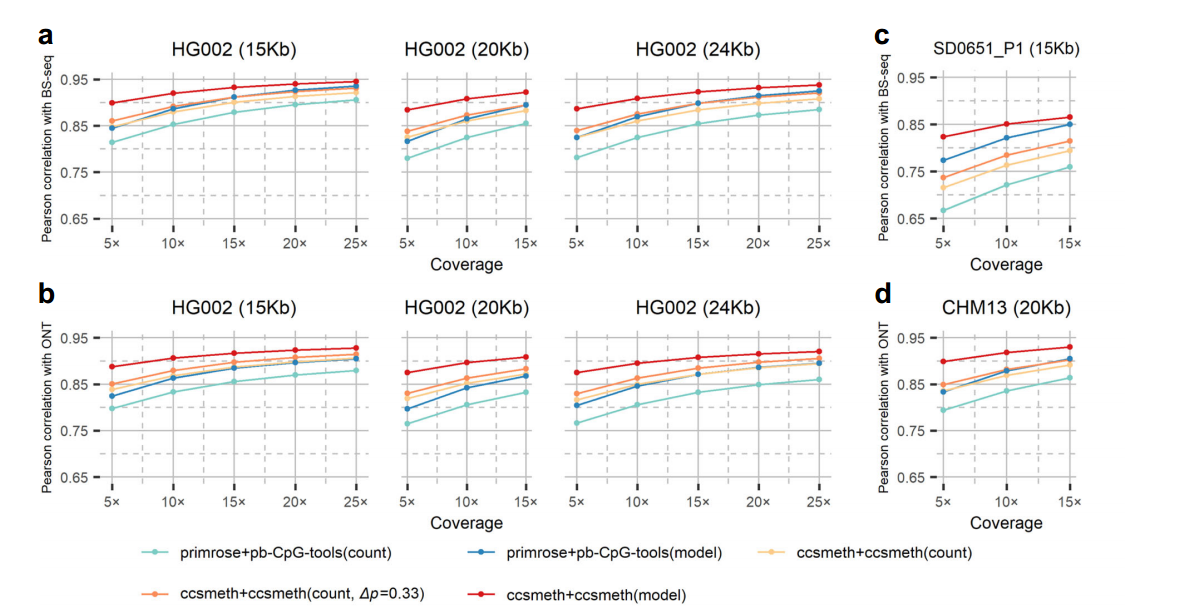
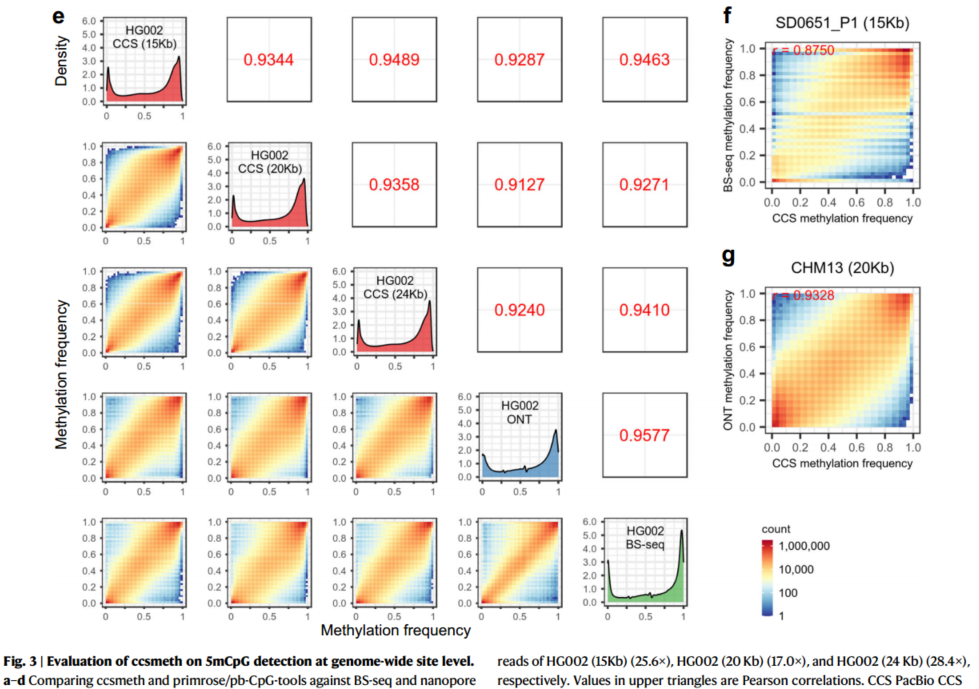
图 3 |在 全 基因组水平评价 ccsmeth 对 5m C p G 检 测 的 影 响 。 a-d 在 HG002、 SD0651_P 1 和 CHM13 CCS reads 不同覆盖范围下, 比较 ccsmeth 和 primrose/pb- cpg 工具与 BS-seq 和纳米孔测序。Δp:甲基化和非甲基化概率之间的差异绝对值。值是 5 次重复测试的平均值。多次重复试验的标准差值见补充表 5-12。e 使用总 CCS 对 ccsmeth 模型模式 与 BS-seq 和纳米孔测序的比较HG002 (15Kb) (25.6×)、HG002 (20 Kb) (17.0×)、HG002 (24 Kb) (28.4×)的reads。上面三角形中的值是皮尔逊相关性。CCS PacBio CCS 测序;ONT 纳米孔测序,BS- seq 亚硫酸酯测序。f使用总计 19.6× SD0651_P 1 (15 Kb) CCS 读取对ccsmeth 模型模式对 BS-seq 的评估。r:皮尔逊相关。g 利用 16.5× CHM13 (20 Kb) CCS 总读取量评估 ccsmet h 模型模式对纳米孔测序的影响。a, b, c 和d 底层的源数据作 为 源 数据文件提供。
基于 PacBio CCS数据的单倍型感知甲基化调用和 ASM 检测
在 ccsmeth 之后,我们进一步开发了一个 38 名为 ccsmethphase 的 Nextflowpipeline, 用于单倍型感知甲基化调用和仅使用 PacBio CCS数据进行 ASM 检测(图 4a, 方法)。在这条管道中, ccsmeth 被用来调用甲基化状态。clair3 被 33 用来命名单核苷酸变异(snv)。然后, 由 Clair3 调用的 snv 被 whatsha46 p 分阶段生成单倍型。DSSis47 用于检测两个单倍型之间的差异甲基化区域(differentially methylated regions,DMRs)。
我们用 71.0× HG002 CCS 读数评估 ccsmethphase(补充表 2)。我们还使用 HG002 BS-seq 数据(补充图 8,补充说明 3)和纳米孔数据(补充图 9, 补充说明 4)来进行 CpG 甲基化相比较。首先, 我们使用 PacBio CCS 数据研究了已知印迹区域的单倍型感知甲基化状态。我们从 Akbari 等 48 人那里得到了 204 个已知印迹区间, 其中有102 个特征明确的印迹区间 49– 53 (方法)。我们比较了 HG002 两种单倍型在每个印迹区间的甲基化差异(方法)。如图 4b 所示, 特征明确证明 ccsmethphase 可以使用 CCS读取准确检测人类基因组中单倍型敏感的甲基化。
评估 PacBio CCS在重复基因组区域的甲基化检测和分阶段
与短读测序技术相比,PacBio CCS 具有更长的读取长度,有望分析人类基因组中更多 CpGs 的甲基化。使用 T2T- chm13 (T2T:端 27 粒到端粒)作为参考基因组, 我们首先评估了 HG002 CCS 读取所覆盖的 CpGs 的数量,特别是在重复的基因组区域:由 RepeatMasker 注释的重复 54,55 基因组元件, 片段重复(SDs)和周围/着丝粒 56 卫-星(cenSats)。57 我们还评估了 HG002 BS-seq 和纳米孔的总读数进行比较(补充表 3)。如图 5a 所示, 使用 15 倍覆盖率的 CCS 读数, 覆盖了 32.85 M(96.9%)的人类 CpGs, 其中 31.33 M CpGs 至少被 5 个读数覆盖。15x CCS reads 覆盖的 CpGs 多于 117.5 x Illumina BS-seq reads 覆盖的 CpGs。当使用所有 71.0×测试 CCS 读取时, 人类基因组中 32.74 M (96.6%) CpGs 被至少5 个映射读取覆盖,这与 65.8×纳米孔读取覆盖的 CpGs 数量几乎相同的印迹区间在两个单倍型之间存在较大的甲基化差异(中位数= 0.53), (图 5a)。在 RepeatMasker 重复序列、SDs 和 cenSats 中,PacBio CCS而其他已知印迹区间中有 22.1%也存在较大的(>0.5)甲基化差异。 CCS 数据得到的已知印迹区间的甲基化差异与 BS-se q 和 nanopore数据得到的甲基化差异高度一致:皮尔逊相关系数分别为 0.8605 和 0.9806(补充图 10 和 11)。我们检查了 SD0651_P1 CCS 数据上已知的印迹区间,得到了一致的结果(补充图 12a)。
然后,我们利用 HG002 测序数据评估 ccsmethphase 在 ASM 检测中的作用。使用 CCS 读取, ccsmethphase 生成 14,390 DMRs。使用 BS-se q 和纳米孔读取相应的管道, 分别产生 2463 和 16250 DMRs。使用 BS- seq 读取产生的 81.4% DMRs 与 CCS 产生的 DMRs的基因组位置非常接近(距离<10 kb), 70.8%的 DMRs 与 CCS 产生的 DMRs 重叠(图 4c)。在使用纳米孔读取产生的 DMRs 中, 68.8%的 DMRs 紧挨着 CCS 产生的 DMRs 的基因组位置, 51.7%的 DMRs与 CCS 产生的 DMRs 重叠(图 4d)。大多数 CCS 产生的 DMRs 也非常接近 HG002 中使用 BS-se q 和纳米孔数据生成的 DMRs 的基因组位置(补充图 13)。从 SD0651_P1 CCS 读取, ccsmethphase 产生 8,183 个 DMRs。在 H G002 和 SD0651 中, 大多数已知的印迹间隔都与 CCS 产生的 DMRs 重叠或靠近(图 4e,补充图 12b 和 14),这也表明 ccsmethphase 对 ASM 检测的能力。我们还利用一个中国家庭的 CCS 数据对 ccsmethphase 的 ASM 检测进行了评估, 其中 HN0641_FA 为父亲, HN0641_MO 为母亲, HN0641_S1 为儿子。结果表明, ccsmephase 不仅可以检测到已知的印迹间隔, 而且可以正确地揭示亲代印迹的模式(补充注 6,补充图 15-18)。
我们进一步比较了 PacBio CCS 数据与 BS-se q 和纳米孔数据检测到的两种单倍型 CpGs 的全基因组位点水平甲基化频率。为了实现这一点, 我们期望一致的单倍型分配(即, 所有母亲 snv 被分配到一个单倍型,所有父亲 snv 被分配到另一个单倍型)。然而,由于参考基因组的 reads 覆盖率不均匀,当使用单个样本的 reads 来分期 snv34 时,我们只能产生离散的单倍型块。因此,我们使用 HG002的 Illumina 三重数据生成的相位 snv 来相位 CCS 和纳米孔读数。我们比较了使用 CCS 读取预测的阶段性 CpGs 的甲基化频率与使用 BS- seq 和纳米孔读取预测的甲基化频率。在母系和父系单倍型中, PacBio CCS与 BS- seq 和纳米孔测序的相关性均>0.93(图 4f, g,补充表 13)。这一结果进一步说明了分别检测到 96.8%、88.4%和 85.3%的 CpGs 甲基化状态。与 BS-se q 相比, RepeatMasker 重复序列、SDs 和 cenSats 中 10.4%、33.6%和 34.7% 的 甲 基化状态 只能分别通过 CCS 检测到( 图 5b)。 在非 repeatmasker 区域,PacBio CCS检测到 96.1%的 CpGs 甲基化状态,比 BS-seq 检测到的 CpGs 甲基化状态高 8.9%(补充图 19a)。
HG002 CCS 序列短于 HG002 纳米孔序列(平均序列长度:18,797 bp vs. 21,933 bp)。然而,PacBio CCS相控的 Cp Gs 数量(即至少有 5 个相控 CCS读段覆盖的 CpGs)并不明显少于纳米孔测序相控的 CpGs 数量:26.97 M vs. 27.66 M(图 5c)。与 BS-se q 相比,PacBio CCS和纳 米孔测序都含有更多的 CpGs。由于读取长度有限, BS-se q 只能检测到人类 CpGs 的 6.71 M 期。PacBio CCS在 RepeatMasker 重复序列、 SDs 和 cen Sats 中分相的 CpGs 分别为 85.4%、60.2%和 46.5%,比 BS-se q 分相的 CpGs 分别高出 63.8%、45.9%和 35.7%(图 5d)。在非 repeatmasker 区域,PacBio CCS 比 BS-seq 多相位 64.4%的 CpGs(补 充图 19b)。值得注意的是, 在 cenSats 中, PacBio CCS 的 Cp Gs 比纳 米孔测序略多, 这可能表明,与纳米孔 R9.4.1 测序相比,高精度的 CCS 测序更适合于 SNV 检测和跨着丝粒区/着丝粒区的甲基化分相。
使用 PacBio CCS预测的重复基因组区域 CpGs 甲基化频率与使用 BS- seq 和纳米孔测序预测的甲基化频率高度相关。 HG002 的 RepeatMasker 重复数、SDs 和 cen Sats 中, PacBio CCS 与 BS- seq 的相关性分别为 0.9540、0.9208 和 0.8822, 与纳米孔测序的相关性分别为 0.9358、0.9087 和 0.8572(补充表 16)。对于 HG002 重复基因组区域的单倍型感知甲基化检测, PacBio CCS 与 BS- seq 和纳米孔测序的相关性也分别为>0.89 和>0.90(补充表 17) 。综上所述, 通过 ccsmeth 和 ccsmethphase, Pac Bio CCS 可以成为重复基因组区域中 5mCpG检测和甲基化相位的全面而准确的技术。
讨论
由于其高准确度的长读长, PacBio CCS在基因组组装 31、SNV检测 33 和结构变异(SV)检测 35 等基因组学研究中得到了越来越广泛的应用。然而, 与纳米孔测序相比, PacBio CCS在 DNA 5m C 检测和甲基化分相中的应用尚未得到充分的研究。在这项研究中, 我们开发并验证了 ccsmeth, 这是一种深度学习方法, 用于从 PacBio CCS 读取中检测 5mcpg。
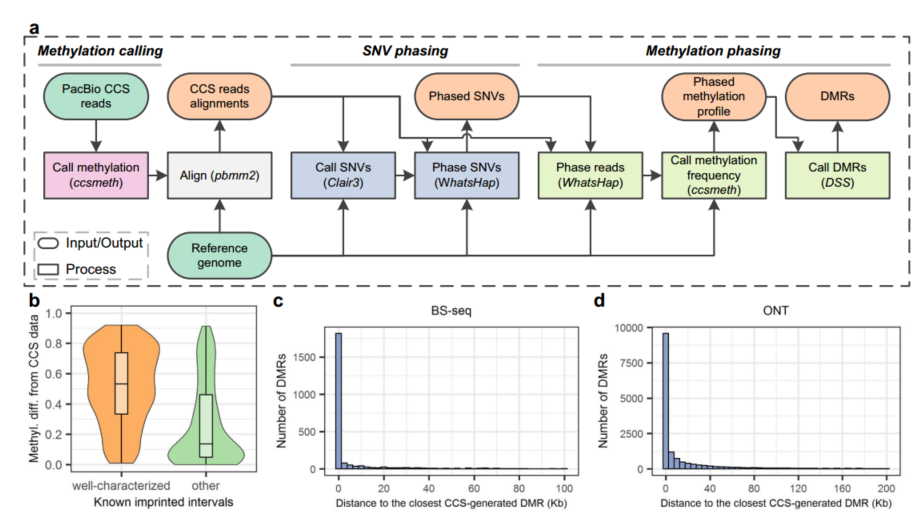
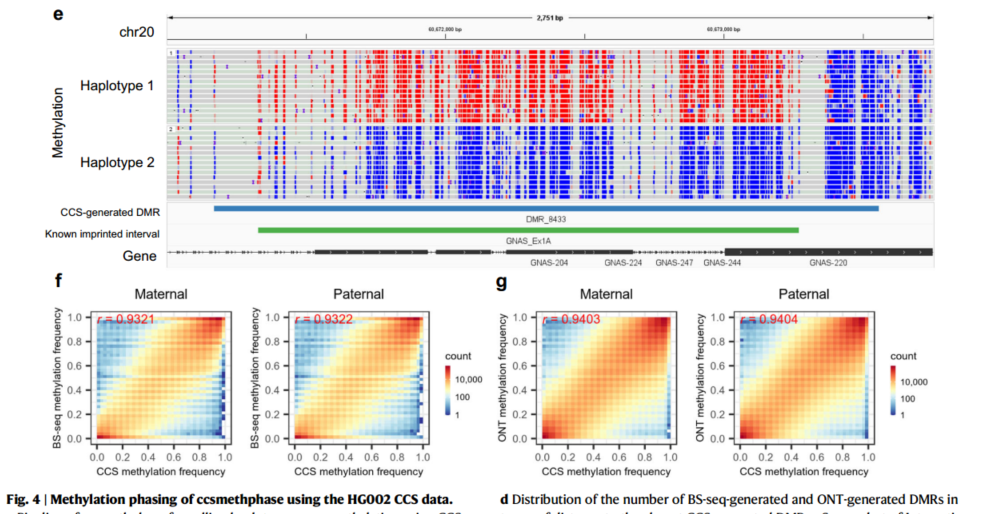
图 4 |使用 HG002 CCS 数据的 ccsmethp h as e 的 甲 基 化 相 位。 使用 CCS 数据调用单倍型感知甲基化的 ccsmethphase 管道。b HG002 两种单倍型 CCS 数据计算的已知印迹区间甲基化差异分布。分析了 102 个“ 良好表征”区间中的 96 个, 以及 102个“ 其他” 区间中的 95 个, 每个单倍型中至少有 5 个 CpGs 被 CCS 读取覆盖。小提琴谱图中的方框分别表示第 50 百分位数(中线)、第 25 百分位数和第 75 百分位数(方框),小于第 25 百分位数的 1.5 倍四分位间距内的最小值和大于第 75 百分位数的 1.5 倍四分位间距内的最大值(须)。c,d bs -seq 生成的 DMR 和 ont 生成的 DMR 的数量与最近的 ccs 生成的 DMR 的距离 的分布。e 在母体印迹基因 GNAS 附近 HG002 的 DMR 上, Integrative Genomics Viewer (chr20:60,671,001-60,673,750)的截图。红点和蓝点分别代表高甲基化概率和 低甲基化概率的 CpGs。f, g PacBio CCS 与 BS- seq 和纳米孔测序在 Illumina 三联体数据中对母、父本单倍型位点水平甲基化频率的比较。甲基。差异, 甲基化差异, r: Pearson 相关, ONT 纳米孔测序。b、 c 和 d 的源数据作为源数据文件提供。
利用 BiGRU 和 attention 机制,我们在 ccsmeth 中设计了两个深度学习模型,分别在读段水平和全基因组位点水平对 5mCp G 进行检测。此外,我们开发了 Nextflow 管道 ccsmethphase,用于甲基化相位和 ASM 检测,仅使用 PacBio CCS读取。我们使用对照(pcr 处理和 m.s si 处理)甲基化数据集、BS-se q 和纳米孔测序系统地评估了多个人体样本的 ccsmeth。ccsmeth 在读码水平和位点水平的 5mC 预测方面优于现有的两种基于 cnn 的方法。对长 CCS读数的评估表明, ccsmeth 达到了
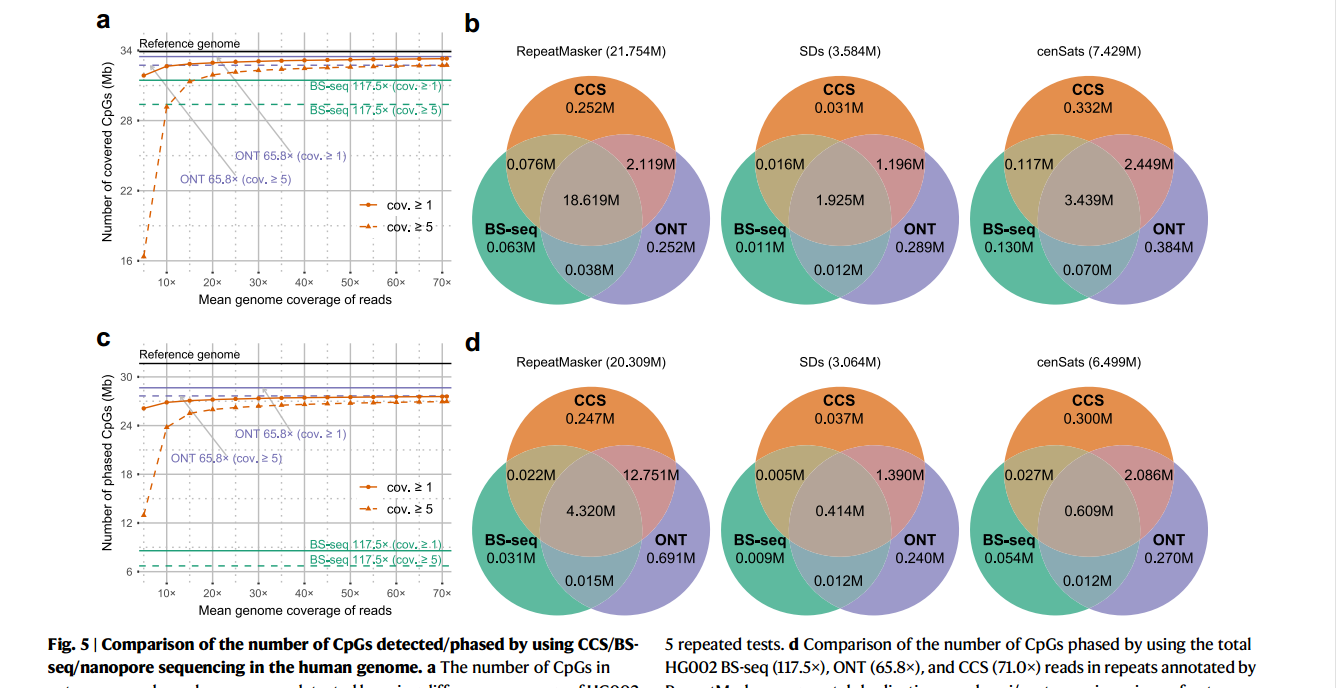
图 5 | CCS/BS - seq/纳 米 孔 测 序在人类基因组中检测 /分相的 CpGs 数 量 比 较。 a 利用 HG002 CCS reads 的差异覆盖率检测常染色体和性染色体中 CpGs 的数量。5×- 70×的值是 5 次重复测试的平均值。b 比较常染色体和性染色体的 HG002 BS-seq序列(117.5×)、ONT 序列(65.8×)和 CCS 序列(71.0×)在 RepeatMasker 标记的重复序列、片段重复和周/着丝粒区检测到的 CpGs 数量。分析了至少 5 个 reads 覆盖的 CpGs。c 使用 HG002 CCS 读段的不同覆盖率来分期常染色体中 CpGs 的数量。 5×-70×的值是的平均值5 次重复测试。d 比较使用 HG002 BS-seq (117.5×)、ONT (65.8×)和 CCS (71.0×)重复序列(RepeatMasker 注释)、片段重复和常染色体周围/着丝粒区进行的 CpGs 数量的比较。分析了至少 5 个阶段读取覆盖的 CpGs。图 a 和 c 的多次重复测试的标准差值见补充表 14-15。子图 b 和 d 中韦恩图标题中的值为T2T-CHM13 基因组相 应 区域的 cpg 总数。浸。覆盖,SDs 片段重复,cenSats 周围/着丝粒卫-星。a 和 c的源数据作为源数据文件提供。
站点级预测的更好性能, 特别是在低覆盖率下。此外, 我们发现 ccsmeth 的站点级模型也可以应用于其他基于 ccsmeth 的方法的读取级结果。实验还表明,随着读取质量和产量的提高(如最近引入的 Revio 系统), PacBio CCS 有潜力完成基因组组装, 检测 SVs, snv 和甲基化使用单个样品的单次测序运行。
纳米孔测序已被证明有助于单倍型 48,PacBio CCS检测到的 DMRs 也可以进一步研究有助于单倍型和基因组组装。
目前, ccsmeth 以及其他使用 PacBio CCS 进行甲基化检测的方法也存在局限性。首先, 尽管 PacBio CCS 读取可以用于检测链特检测非人类物种的甲基化在遗传学和表观遗传学领域也很重要。 异性甲基化, 但所有这些方法都只考虑对称甲基化, 并结合来自两在本研究中, 我们进一步对斑马鱼 DNA 样本并行进行 PacBio CCS和 BS-seq, 以评估非人类数据的 ccsmeth(方法)。我们使用预训练的 ccsmeth 模型从斑马鱼样本的 CCS 读取中检测 5mCpG 甲基化, 并将结果与 BS- seq 进行比较。我们还使用报春花进行比较, 从 CCS 读取中检测到 5mCpG 甲基化。结果表明, 与报春花相比, ccsmeth 与 BS-se q 的相关性更高(0.8463 vs. 0.8292),这表明 ccsmeth 在检测非人类数据的甲基化方面具有鲁棒性(Supplementary Fig. 20)。
为了评估 ccsmethphase,我们分别使用 BS- seq 和纳米孔测序设计了另外两个甲基化相的管道, 这些管道参考了之前的一些研究 34,58,59。使用 ccsmethphase 的全基因组甲基化分相结果与 BS-seq和纳米孔测序高度一致, 包括在已知的印迹间隔内。对甲基化相的评估表明, PacBio CCS 在甲基化相上与纳米孔测序相当。这两种长读技术都可以检测到比 BS-seq 更多的单倍型感知 Cp Gs 的甲基化状态。而单倍型之间的 DMRs 检测
个 DNA 链的特征来预测甲基化状态。因此, 这些方法不能检测半甲基化的 cpg, 也不能检测非 cpg 中的 5mC 或其他不具有 60 对称甲基化模式的 DNA 修饰(如 6mA)。我们重新设计了 ccsmeth 模型, 使用长 CCS 读取来调用链特异性甲基化(补充图 21a)。该链特异性甲基化模型在 HG002 15 Kb 数据集中的读级精度达到 0.85(补充图 21b)。然而,与对称-甲基化模型相比, 由于亚读深度有限,该模型的性能明显降低(补充图 21b, c)。其次,位点级模型的设计是否可以直接应用于非 cpg 5mc 的检测等修改尚未得到验证。然而, 通过生成更多用于其他修改的地面真实数据集, 并根据其他修改的相应模式重新设计 ccsmeth 模型, 我们相信这些局限性在未来的研究中可能都能得到解决。
综上所述, ccsmeth 和 ccsmeth- phase 与 PacBio CCS 一起可以成为全基因组 5mCp G 检测和甲基化分相的良好应用方法。我们期望我们提出的方法将有助于单倍型甲基化机制的分析以及其他修饰的检测。
| 阿里云国内75折 回扣 微信号:monov8 |
| 阿里云国际,腾讯云国际,低至75折。AWS 93折 免费开户实名账号 代冲值 优惠多多 微信号:monov8 飞机:@monov6 |